Por: Enrique Blanco
Un enfoque técnico-regulatorio para la dirección de proyectos de remediación, transferencia de tecnología, ampliación y construcción de nuevas plantas farmacéuticas bajo los marcos de FDA, EMA y Cofepris, centrado en la protección del paciente.
Resumen
La gestión de proyectos farmacéuticos representa uno de los entornos más complejos de la dirección de proyectos, al conjugar las exigencias del Project Management Body of Knowledge (PMBOK®) del PMI con los marcos regulatorios internacionales de Food & Drug Administration (FDA), European Medicines Agency (EMA) y la Comisión Federal para la Protección contra Riesgos Sanitarios (Cofepris), los principios de gestión del riesgo de calidad de ICH Q9 (R1), las guías de transferencia de tecnología de la PDA Technical Report No. 65 y los estándares de ingeniería de ISPE. El presente artículo desarrolla un marco integrado para la planificación, ejecución y control de proyectos de remediación GMP, ampliación de capacidad productiva y construcción de nuevas plantas farmacéuticas, incorporando la perspectiva centrada en el paciente como eje fundamental de las decisiones técnicas y de riesgo.
01. INTRODUCCIÓN
La industria farmacéutica como entorno de alta complejidad proyectual
La industria farmacéutica exige que cada proyecto —desde la remediación de una sala limpia hasta la construcción greenfield de una nueva planta estéril— sea ejecutado bajo una lógica dual: la eficiencia del proyecto (tiempo, costo, alcance) y la garantía regulatoria y de calidad (cumplimiento de BPF/GMP, protección del paciente). Esta dualidad eleva sustancialmente la complejidad de la gestión proyectual respecto a otros sectores industriales1,2.
El fracaso en proyectos farmacéuticos no se limita a sobrecostos o retrasos, puede derivar en desabasto de medicamentos esenciales, observaciones de inspección (Form FDA 483, Warning Letters, cartas Cofepris) y, en el peor de los casos, en daño directo al paciente. Por ello, la integración metodológica es imprescindible3.

02. PMBOK EN EL ENTORNO FARMACÉUTICO
Aplicación del PMBOK® 7.ª Edición en Proyectos Farmacéuticos
La séptima edición del PMBOK® del Project Management Institute (PMI) migra de un enfoque de procesos hacia un modelo basado en principios y dominios de desempeño, lo cual resulta particularmente alineado con la visión regulatoria farmacéutica basada en riesgo. Los doce principios del PMBOK® 7 encuentran correspondencia directa con los requerimientos de las autoridades sanitarias4.
2.1. Grupos de Procesos aplicados a la manufactura farmacéutica
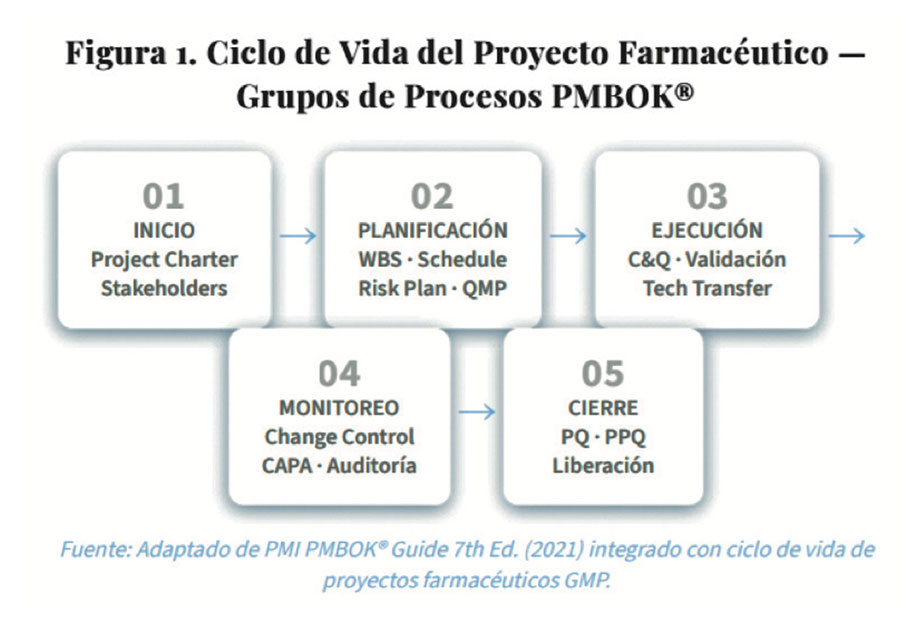
2.2. Acta de Constitución del Proyecto (Project Charter) con componentes regulatorios
En el entorno farmacéutico regulado, el Project Charter debe incorporar elementos adicionales a los estándares del PMI. La justificación del negocio debe vincular el proyecto con el mantenimiento de licencias sanitarias (NOM-059-SSA1, 21 CFR Parts 210/211, EU GMP Annex 1), la continuidad de suministro y la protección de los derechos del paciente5.
Componentes regulatorios obligatorios del Project Charter Farmacéutico: (1) Número de autorización sanitaria impactada, (2) Evaluación preliminar de riesgo a paciente (ICH Q9), (3) Matriz de jurisdicciones regulatorias aplicables, (4) Requisitos de notificación o aprobación previa a autoridades (Prior Approval Supplement – FDA, Variación Tipo II – EMA, Aviso de Modificación – Cofepris) y (5) Plan de continuidad de suministro durante el proyecto.
2.3. Estructura de desglose del trabajo (WBS) para Proyectos GMP
La WBS en proyectos farmacéuticos debe reflejar no solo los entregables de ingeniería/construcción, sino también los paquetes de trabajo regulatorios y de calidad. Una WBS bien estructurada permite la trazabilidad entre entregables físicos y documentos GMP requeridos por la FDA, EMA y Cofepris6.
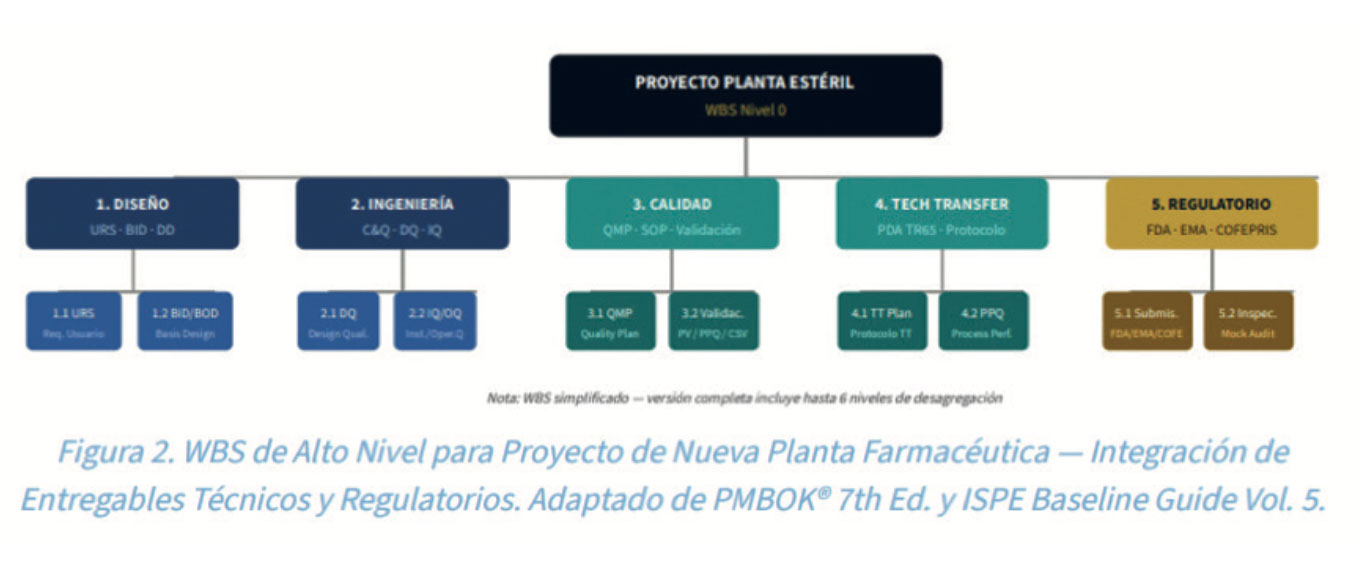
2.4. Gestión del Cronograma y Ruta Crítica en proyectos regulados
El método de la ruta crítica (CPM) y la gestión del valor ganado (EVM) son herramientas esenciales; sin embargo, en proyectos farmacéuticos existe una ruta crítica regulatoria paralela que debe monitorearse con igual rigor: los tiempos de revisión y aprobación por parte de la FDA, EMA o Cofepris pueden superar los 12 meses para suplementos de aprobación previa (PAS), lo que puede convertirse en el determinante del plazo total del proyecto, independientemente del desempeño de la construcción7.
 Ruta Crítica Regulatoria. En proyectos con impacto en registros sanitarios (CMC changes, site changes), el director de proyecto debe incorporar los tiempos de respuesta regulatoria como actividades formales en el cronograma. FDA PDUFA goals establecen 10 meses para PAS de medicamentos NDA; EMA puede requerir hasta 90 días para variaciones Tipo II; Cofepris hasta 180 días hábiles para modificaciones que requieren autorización previa según la NOM-073-SSA1-2015.
Ruta Crítica Regulatoria. En proyectos con impacto en registros sanitarios (CMC changes, site changes), el director de proyecto debe incorporar los tiempos de respuesta regulatoria como actividades formales en el cronograma. FDA PDUFA goals establecen 10 meses para PAS de medicamentos NDA; EMA puede requerir hasta 90 días para variaciones Tipo II; Cofepris hasta 180 días hábiles para modificaciones que requieren autorización previa según la NOM-073-SSA1-2015.
03. GESTIÓN DEL RIESGO
ICH Q9(R1): Gestión del Riesgo de Calidad centrada en el paciente
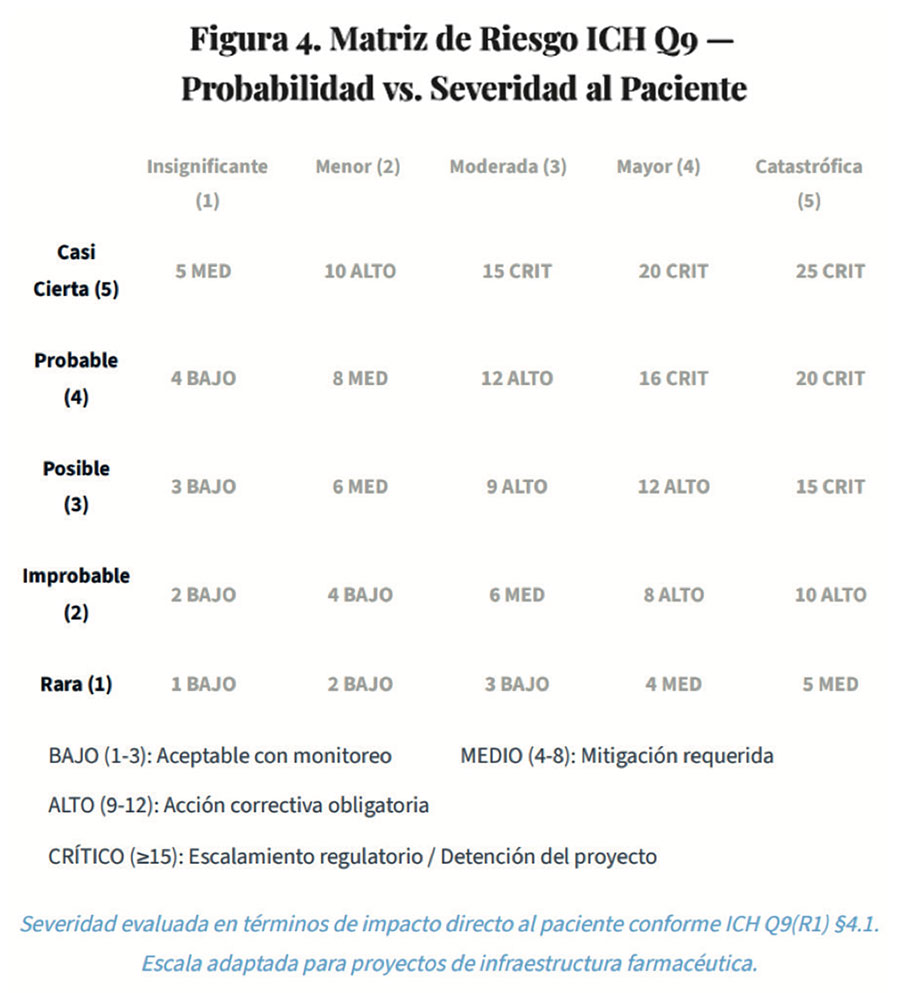
La revisión 1 de ICH Q9, publicada en 2023, fortalece explícitamente la perspectiva del paciente como eje central de todas las evaluaciones de riesgo de calidad. Este principio tiene implicaciones directas en cómo se priorizan los riesgos en los proyectos farmacéuticos: no basta con evaluar el riesgo para el negocio o la operación; la pregunta fundamental es "¿cómo impacta este riesgo en la seguridad, eficacia y disponibilidad del medicamento para el paciente?"8.
3.1. Proceso de Gestión del Riesgo de Calidad (QRM) — ICH Q9(R1)
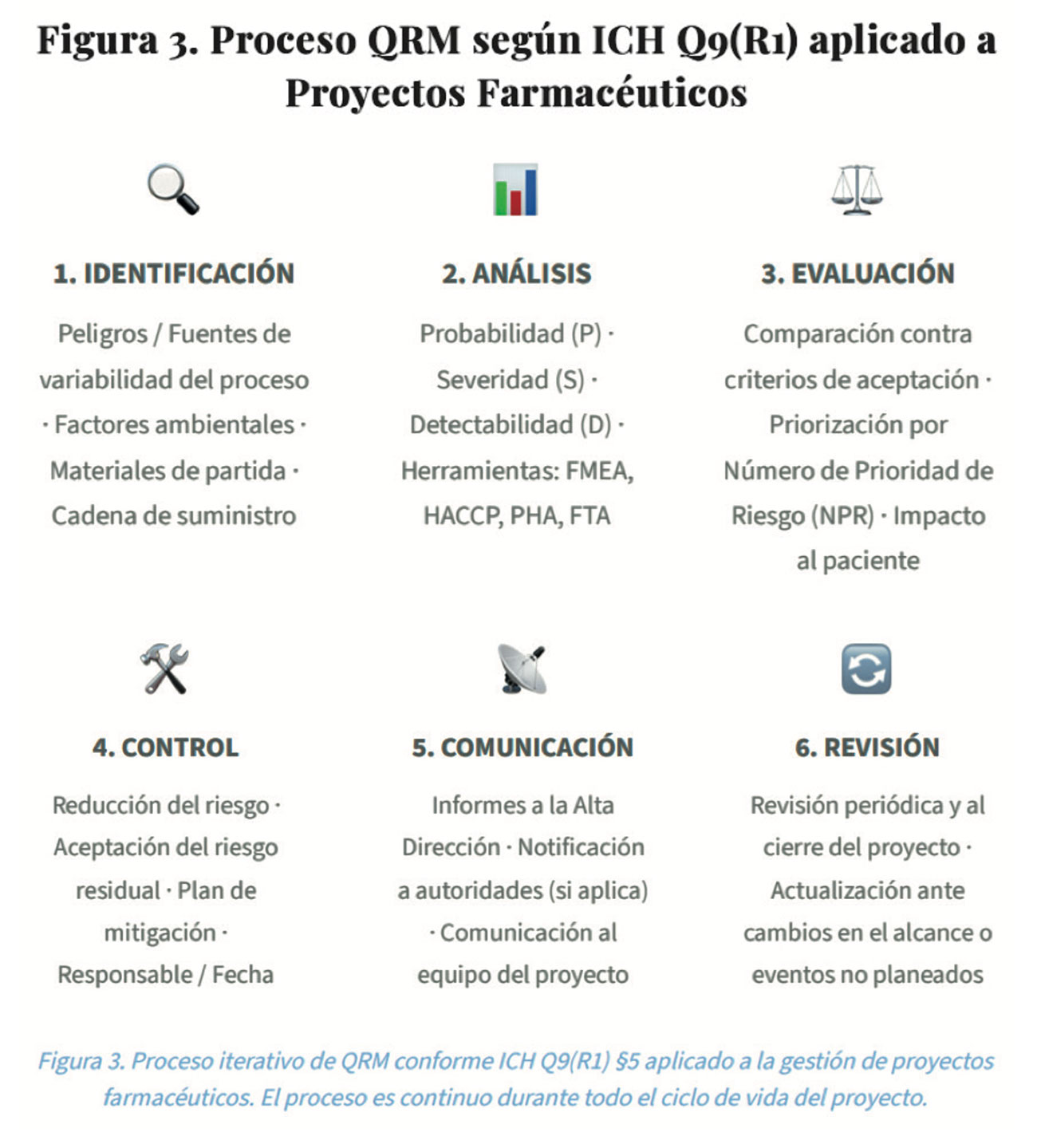
3.2. Perspectiva del paciente en la Evaluación de Riesgo de Proyectos
ICH Q9(R1) establece que la severidad del daño debe evaluarse primariamente en términos del impacto potencial en el paciente, no en el proceso o el negocio. En el contexto de proyectos, esto significa que cualquier cambio en instalaciones, equipos o procesos debe ser evaluado considerando8,9:

3.3. Matriz de Riesgo Integrada para proyectos farmacéuticos
04. TRANSFERENCIA DE TECNOLOGÍA
PDA Technical Report No. 65: Gestión del Riesgo de Tecnología en Transferencias de Proceso
El Technical Report No. 65 de la Parenteral Drug Association (PDA), "Technology Transfer" establece un marco estructurado para la transferencia de procesos farmacéuticos entre sitios de manufactura (site-to-site), de investigación y desarrollo hacia producción comercial (R&D-to-commercial) y transferencias entre organizaciones (inter-company). La PDA TR65 define la transferencia de tecnología como un proceso lógico y documentado de transmisión de conocimiento, experiencia e información desde la unidad cedente (sending unit, SU) hacia la unidad receptora (receiving unit, RU)10.
4.1 Fases de Transferencia según PDA TR65
- Fase 1 — Pre-transferencia
Evaluación de Factibilidad y Brecha Tecnológica (Gap Assessment)
Análisis comparativo entre las capacidades técnicas de la SU y la RU. Incluye evaluación de equipos, sistemas de soporte crítico (agua para inyección, HVAC, sistemas de gases), capacidades analíticas y estado de validación. La PDA TR65 recomienda el uso de herramientas QRM (FMEA, análisis de brechas) para identificar riesgos tecnológicos antes de comprometer recursos. Identificación de Critical Quality Attributes (CQAs) y Critical Process Parameters (CPPs) del proceso a transferir10,11.
- Fase 2 — Planificación
Desarrollo del Plan Maestro de Transferencia de Tecnología (TTMP)
El TTMP define el alcance, los criterios de aceptación, los roles y responsabilidades (RACI), el cronograma y el plan de gestión del riesgo específico para la transferencia. Debe contemplar la estrategia de calificación de equipos en la RU (DQ/IQ/OQ/PQ conforme ISPE Baseline), los protocolos analíticos de transferencia de métodos (conformidad con USP <1224> y ICH Q2(R2)) y los requisitos de notificación regulatoria asociados10,12.
- Fase 3 — Ejecución
Manufactura de Lotes de Demostración y Calificación de Proceso
Producción de lotes de demostración en la RU bajo supervisión conjunta SU-RU. Validación de limpieza y contaminación cruzada (EU GMP Annex 15, FDA Process Validation Guidance 2011). Transferencia de métodos analíticos con estudios de exactitud, precisión, linealidad y especificidad. Documentación de desviaciones y acciones correctivas durante los lotes de calificación10,13.
- Fase 4 — Validación
Calificación de Desempeño del Proceso (PPQ — Process Performance Qualification)
Ejecución del protocolo PPQ conforme FDA Process Validation Guidance (2011) y EMA GL on process validation. Número estadísticamente justificado de lotes (mínimo 3, aunque la justificación basada en riesgo puede requerir más). Demostración de capacidad de proceso (Cpk ≥ 1.33 para características críticas). Revisión de datos de CPPs vs. CQAs mediante herramientas de análisis estadístico (SPC, DoE confirmatorio)13,14.
- Fase 5 — Cierre y liberación comercial
Informe de Transferencia y Aprobación Regulatoria
Emisión del Informe Final de Transferencia de Tecnología con conclusiones sobre el cumplimiento de criterios de aceptación. Notificación o solicitud de aprobación a autoridades regulatorias según la naturaleza del cambio (Annual Report, CBE-30, PAS para FDA; Type IA, IB o II para EMA; Modificación con o sin autorización previa para COFEPRIS). Actualización del Dossier de Registro (CTD/eCTD Módulo 3)10,15.
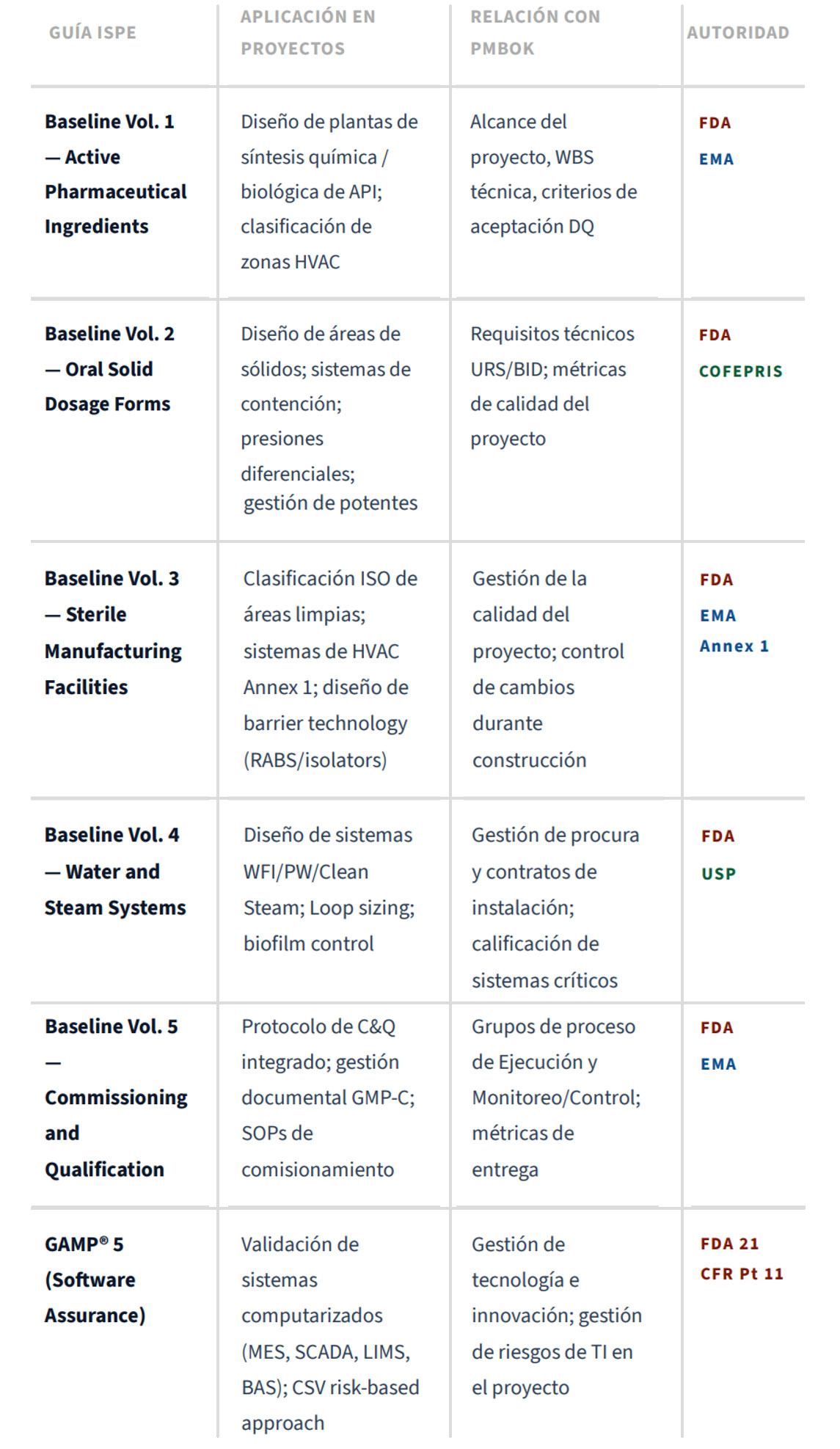
4.1 Guías ISPE para Proyectos de Instalaciones Farmacéuticas
La International Society for Pharmaceutical Engineering (ISPE) proporciona a través de sus Baseline® Pharmaceutical Engineering Guides los estándares de referencia para el diseño y la construcción de instalaciones farmacéuticas. Los volúmenes más relevantes para proyectos de infraestructura son16:
05. INTEGRACIÓN METODOLÓGICA
Cruce Metodológico: PMBOK® vs. PDA Technical Report 65
La integración de las metodologías PMBOK® y PDA TR65 no es simplemente un ejercicio académico es una necesidad operativa para los directores de proyectos farmacéuticos que deben satisfacer simultáneamente las expectativas de sus sponsors corporativos (eficiencia, costo, tiempo) y las exigencias de las autoridades regulatorias (integridad, trazabilidad, cumplimiento GMP). La siguiente tabla establece la correspondencia sistemática entre ambos marcos4,10.
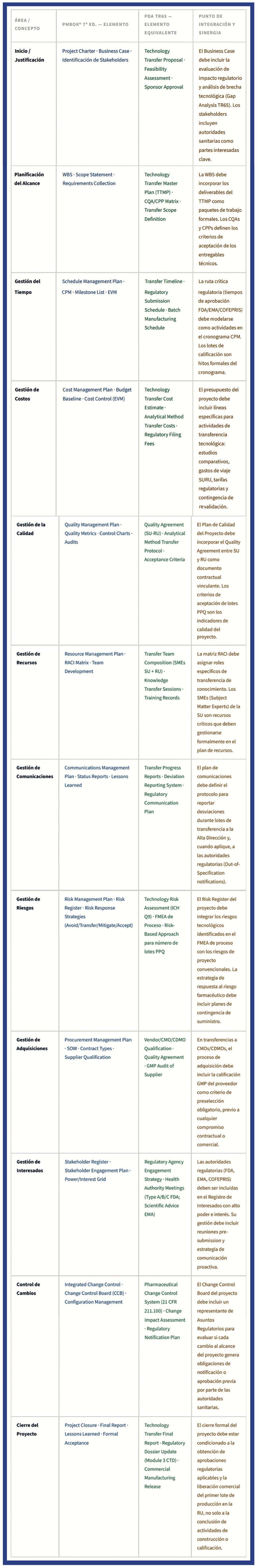 Principio de Integración Clave. La metodología PMBOK® provee la estructura de gobierno y gestión del proyecto; la PDA TR65 provee el contenido técnico y los criterios de aceptación específicos del dominio farmacéutico. Ninguno de los dos marcos por sí solo es suficiente para gestionar exitosamente un proyecto de transferencia tecnológica farmacéutica. El director de proyecto farmacéutico debe ser competente en ambos.
Principio de Integración Clave. La metodología PMBOK® provee la estructura de gobierno y gestión del proyecto; la PDA TR65 provee el contenido técnico y los criterios de aceptación específicos del dominio farmacéutico. Ninguno de los dos marcos por sí solo es suficiente para gestionar exitosamente un proyecto de transferencia tecnológica farmacéutica. El director de proyecto farmacéutico debe ser competente en ambos.
06. MARCO REGULATORIO
Requisitos Regulatorios: FDA, EMA y Cofepris en Proyectos Farmacéuticos
6.1 Food and Drug Administration (FDA) — Estados Unidos
La FDA regula los cambios en procesos, instalaciones y equipos de manufactura farmacéutica a través del sistema de Chemistry, Manufacturing, and Controls (CMC) Post-Approval Changes, regulado bajo 21 CFR Part 314 (NDA) y 21 CFR Part 601 (BLA). El nivel de documentación requerido (Annual Report, CBE-30 o Prior Approval Supplement) depende del potencial impacto del cambio en la identidad, concentración, calidad, pureza o potencia del producto17.

6.2 European Medicines Agency (EMA) — Unión Europea
El sistema europeo de gestión de variaciones (Regulation EC No. 1234/2008, actualizado por Regulation EU 2022/2058) clasifica los cambios post-aprobación en variaciones Tipo IA (menores, notificación inmediata), Tipo IB (notificación, implementación tras 30 días sin objeción) y Tipo II (aprobación previa requerida). Las guías EMA sobre process validation y el EU GMP Annex 1 (2022) para manufactura aséptica establecen los estándares técnicos aplicables18,19.
EU GMP Annex 1 (2022) — Impacto en Proyectos de Plantas Estériles:
La revisión de 2022 del Annex 1 introduce requerimientos significativamente más estrictos para sistemas de contaminación de partículas, Contamination Control Strategy (CCS), integración de tecnología RABS e isolators, y monitoreo ambiental continuo. Todos los proyectos de plantas estériles o expansiones de capacidad deben ser evaluados contra estos nuevos requerimientos como parte del alcance del proyecto, independientemente de si el sitio es nuevo o existente.
6.3 Cofepris — México
La Comisión Federal para la Protección contra Riesgos Sanitarios (Cofepris) regula los cambios post-autorización mediante la NOM-073-SSA1-2015 (estabilidad de fármacos) y los lineamientos de modificaciones a registros sanitarios. El sistema mexicano distingue entre modificaciones que requieren autorización previa, notificación previa y notificación posterior. La implementación del sistema de Buenas Prácticas de Fabricación en México se alinea progresivamente con los estándares del Esquema de Cooperación de Inspección Farmacéutica (PIC/S), del cual México es miembro desde 202120.
- Alineación PIC/S de la Cofepris. La adhesión de México al PIC/S implica que las inspecciones de la Cofepris se realizan bajo los estándares armonizados con FDA y EMA, incluyendo el enfoque de inspección basado en riesgo (RBI). Los proyectos de expansión o remediación deben considerar que una inspección de la Cofepris post-proyecto evaluará el sistema de calidad completo, no solo los cambios realizados.
07. C&Q / VALIDACIÓN
Comisionamiento, Calificación y Validación (C&Q/V) como Entregables del Proyecto
El proceso de Comisionamiento y Calificación (C&Q) representa la interfaz crítica entre la fase de construcción (dominio del proyecto) y la fase de operación regulada (dominio GMP). La ISPE Baseline Guide Vol. 5 y las guías ICH Q8-Q10 establecen que el C&Q debe ser un proceso continuo e integrado, no una actividad de último momento12,16.
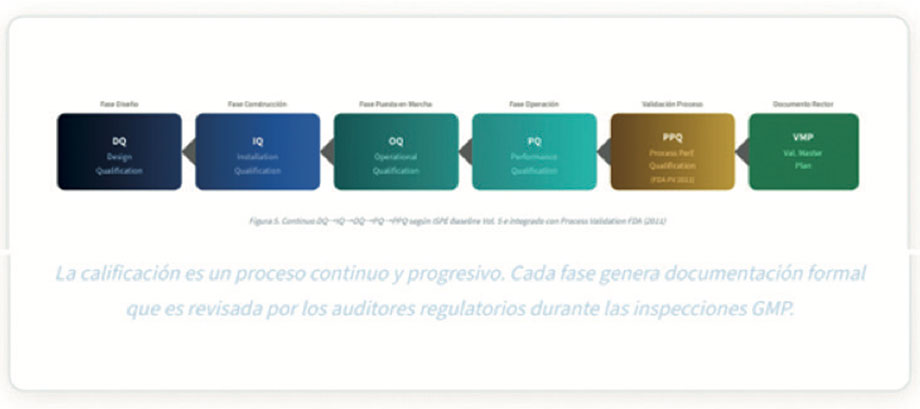
7.1 Validation Master Plan (VMP) como Entregable Clave del Proyecto
El VMP es el documento rector que establece la estrategia de validación para toda la instalación o proyecto. Su desarrollo debe iniciarse en la fase de diseño (con el DQ como input) y debe ser aprobado por el director de Calidad antes del inicio de la construcción o modificación. Para inspecciones FDA, EMA y Cofepris, la ausencia o deficiencia del VMP es frecuentemente citada como observación mayor (Major Finding en inspecciones EMA)21.
Contenido Mínimo del VMP según EU GMP Annex 15 y FDA Guidance: (1) Descripción de la instalación/proceso/sistema objeto de validación, (2) Estructura organizacional del equipo de validación, (3) Lista de sistemas y equipos clasificados como GMP-críticos, (4) Programa de revalidación periódica, (5) Gestión del change control post-validación, (6) Metodología estadística para determinación del número de corridas, (7) Criterios de aceptación globales y por sistema, (8) Manejo de desviaciones durante la validación.
08. TIPOS DE PROYECTOS
8.1 Proyectos de Remediación GMP (Remediation Projects)
Los proyectos de remediación representan el escenario de mayor urgencia y riesgo en la industria farmacéutica. Se activan típicamente tras la recepción de un Warning Letter (FDA), una deficiencia mayor de inspección (EMA) o una medida de seguridad sanitaria (Cofepri). Su característica principal es la presión temporal extrema combinada con alta visibilidad regulatoria22.
 Gestión de Crisis en Proyectos de Remediación. El director de proyecto de remediación debe gestionar simultáneamente: (1) el plan de acción correctiva comprometido con la autoridad regulatoria (CAPA comprometido), (2) la continuidad de suministro del portafolio de productos (para evitar shortage notification ante FDA bajo 21 CFR 314.81(b)(3)(iii)), (3) la comunicación periódica con la autoridad regulatoria sobre el avance, (4) el sistema de change control interno que documente cada acción remediadora. El fracaso en cualquiera de estos frentes puede resultar en escalamiento regulatorio (Import Alert, Consent Decree).
Gestión de Crisis en Proyectos de Remediación. El director de proyecto de remediación debe gestionar simultáneamente: (1) el plan de acción correctiva comprometido con la autoridad regulatoria (CAPA comprometido), (2) la continuidad de suministro del portafolio de productos (para evitar shortage notification ante FDA bajo 21 CFR 314.81(b)(3)(iii)), (3) la comunicación periódica con la autoridad regulatoria sobre el avance, (4) el sistema de change control interno que documente cada acción remediadora. El fracaso en cualquiera de estos frentes puede resultar en escalamiento regulatorio (Import Alert, Consent Decree).
La metodología de proyectos de remediación exitosos incluye invariablemente: diagnóstico sistémico del sistema de calidad (no solo de los hallazgos de inspección), definición de causas raíz con herramientas como 5-Why o Ishikawa, plan de remediación con categorización de acciones por plazo (inmediato, corto, mediano, largo), y un sistema robusto de monitoreo de efectividad de las acciones implementadas (E ectiveness Check) 22,23.
8.2 Proyectos de Ampliación de Capacidad
Los proyectos de expansión de capacidad (debottlenecking, scale-up, adición de líneas de producción) requieren una gestión cuidadosa del impacto regulatorio de los cambios. Bajo el concepto de Quality by Design (QbD) de ICH Q8(R2), las ampliaciones dentro del Design Space aprobado no requieren necesariamente aprobación regulatoria previa, lo que representa una ventaja competitiva significativa cuando el espacio de diseño fue correctamente establecido durante el desarrollo24.
8.3 Nuevas Plantas Farmacéuticas (Greenfield y Brownfield)
La construcción de nuevas instalaciones farmacéuticas (greenfield) o la conversión de instalaciones existentes (brownfield) representa el proyecto de mayor complejidad y mayor inversión de capital en el sector. Estos proyectos típicamente involucran inversiones de entre 50 millones de dólares (mdd) (planta de sólidos mediana) y 1,500 mdd (planta biofarmacéutica de última generación), con horizontes de ejecución de tres a siete años25.
- Proyecto Greenfield
Construcción desde cero en terreno nuevo. Mayor flexibilidad de diseño para optimización GMP. Requiere proceso completo de permisos (uso de suelo, impacto ambiental, licencia sanitaria de construcción). Inversión mayor pero menor riesgo de interferencia con producción existente. Pre-licencia Cofepris requerida antes del inicio de construcción.
- Proyecto Brownfield
Modificación o expansión de instalaciones existentes en operación. Mayor riesgo de contaminación cruzada durante construcción (requerimiento de planes de segregación y protocolos de construcción GMP). Impacto regulatorio inmediato sobre licencias sanitarias vigentes. Plan de continuidad de producción obligatorio durante las obras.
09. CONCLUSIONES
La gestión exitosa de proyectos en la industria farmacéutica requiere la integración disciplinada de múltiples marcos metodológicos y regulatorios. El presente artículo ha demostrado que PMBOK®, ICH Q9(R1), PDA TR65, ISPE Baseline Guides y los marcos regulatorios de FDA, EMA y COFEPRIS no son marcos competidores sino complementarios, que juntos forman un sistema de gestión proyectual robusto y defensible ante cualquier autoridad sanitaria.
Los puntos críticos de integración identificados son: (1) la incorporación de la perspectiva del paciente como criterio primario de evaluación de riesgos del proyecto (ICH Q9(R1)); (2) la inclusión de la ruta crítica regulatoria en el cronograma del proyecto como actividad formal (no como reserva de gestión); (3) la construcción del Project Charter con elementos regulatorios específicos que vinculen el proyecto con los registros sanitarios impactados; (4) el cierre formal del proyecto condicionado a la liberación comercial del primer lote, no solo a la conclusión de actividades de infraestructura.
El director de proyecto farmacéutico del siglo XXI debe poseer una competencia híbrida: la disciplina metodológica del Project Management Professional (PMP) combinada con el conocimiento técnico del entorno regulatorio farmacéutico. Esta combinación, junto con la experiencia práctica en la gestión de las complejidades únicas de la industria farmacéutica (control de cambios regulatorio, gestión de desabasto, calificación y validación), define el perfil del profesional que puede agregar valor diferencial en proyectos de alta consecuencia26.
Referencias — Formato Vancouver:
- Project Management Institute. A Guide to the Project Management Body of Knowledge (PMBOK® Guide). 7th ed. Newtown Square, PA: PMI; 2021.
- García-Arieta A, Gordon J. Bioequivalence requirements in the European Union: critical discussion. AAPS J. 2012;14(4):738–48. doi:10.1208/s12248-012-9382-1
- International Council for Harmonisation. ICH Q10: Pharmaceutical Quality System. Geneva: ICH; 2008.
- Project Management Institute. PMBOK® Guide — Seventh Edition and The Standard for Project Management. Newtown Square, PA: PMI; 2021.
- U.S. Food and Drug Administration. 21 CFR Parts 210 and 211 — Current Good Manufacturing Practice Regulations for Finished Pharmaceuticals. Silver Spring, MD: FDA; 2022.
- International Society for Pharmaceutical Engineering. ISPE Baseline® Pharmaceutical Engineering Guide, Volume 5: Commissioning and Qualification. 2nd ed. Bethesda, MD: ISPE; 2019.
- U.S. Food and Drug Administration. Guidance for Industry: Changes to an Approved NDA or ANDA. Silver Spring, MD: FDA; 2004.
- International Council for Harmonisation. ICH Q9(R1): Quality Risk Management. Geneva: ICH; 2023.
- Nasr MM, Krsplits H, Iser R, et al. New and revised ICH guidelines: a framework for pharmaceutical quality. Pharm Technol. 2017;41(3):42–8.
- Parenteral Drug Association. PDA Technical Report No. 65: Technology Transfer. Bethesda, MD: PDA; 2014.
- International Council for Harmonisation. ICH Q8(R2): Pharmaceutical Development. Geneva: ICH; 2009.
- International Society for Pharmaceutical Engineering. ISPE Baseline® Pharmaceutical Engineering Guide, Volume 3: Sterile Manufacturing Facilities. 2nd ed. Bethesda, MD: ISPE; 2011.
- U.S. Food and Drug Administration. Guidance for Industry: Process Validation — General Principles and Practices. Silver Spring, MD: FDA; 2011.
- European Medicines Agency. Guideline on Process Validation for Finished Products — Information and Data to be Provided in Regulatory Submissions (EMA/CHMP/CVMP/QWP/BWP/70278/2012). Amsterdam: EMA; 2016.
- International Council for Harmonisation. ICH M4Q(R1): The Common Technical Document for the Registration of Pharmaceuticals for Human Use — Quality. Geneva: ICH; 2002.
- International Society for Pharmaceutical Engineering. ISPE Baseline® Pharmaceutical Engineering Guide, Volume 1: Active Pharmaceutical Ingredients. 2nd ed. Bethesda, MD: ISPE; 2007.
- U.S. Food and Drug Administration. Guidance for Industry: SUPAC-SS: Nonsterile Semisolid Dosage Forms — Scale-Up and Post-Approval Changes. Silver Spring, MD: FDA; 1997.
- European Commission. Commission Regulation (EC) No 1234/2008 concerning the examination of variations to the terms of marketing authorisations for medicinal products for human use and veterinary medicinal products. Brussels: EU; 2008.
- European Commission. EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use, Annex 1: Manufacture of Sterile Medicinal Products. Brussels: EC; 2022.
- Comisión Federal para la Protección contra Riesgos Sanitarios. NOM-073-SSA1-2015, Estabilidad de Fármacos y Medicamentos. Ciudad de México: Cofepris; 2015.
- European Commission. EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use, Annex 15: Qualification and Validation. Brussels: EC; 2015.
- U.S. Food and Drug Administration. Compliance Policy Guidance 7356.002F — Drug Manufacturing Inspections. Silver Spring, MD: FDA; 2020.
- International Council for Harmonisation. ICH Q12: Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management. Geneva: ICH; 2019.
- International Council for Harmonisation. ICH Q8(R2): Pharmaceutical Development. Geneva: ICH; 2009.
- International Society for Pharmaceutical Engineering. Good Practice Guide: Technology Transfer. Bethesda, MD: ISPE; 2003.
- Blanco E. Solid Dosage Form Product Transfer — Practical Case, Risk Assessment Approach [Poster presentation]. PDA Annual Meeting; 2012.
- Blanco E. Raw material contamination: a case study. J GXP Compliance. 2013.
- United States Pharmacopeia. USP Transfer of Analytical Procedures. Rockville, MD: USP; 2021.
- International Council for Harmonisation. ICH Q2(R2): Validation of Analytical Procedures. Geneva: ICH; 2022.
- Parenteral Drug Association. PDA Technical Report No. 44: Quality Risk Management for Aseptic Processes. Bethesda, MD: PDA; 2008.